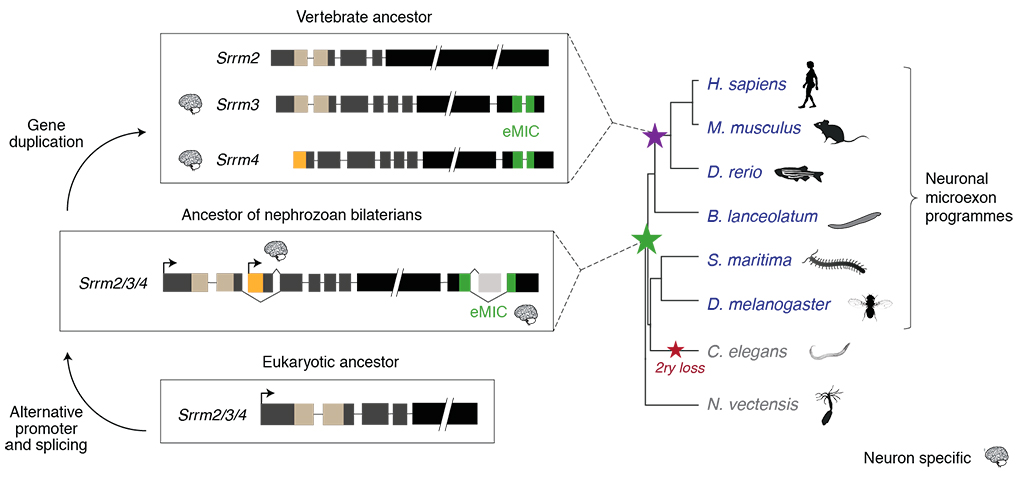
The mechanisms by which entire programs of gene regulation arose are poorly understood, and very rarely can be traced back to singular genomic novelties (e.g. the appearance of a specific regulator or protein domain). In this study, we elucidated the mechanism underlying the emergence of a particularly intriguing and evolutionarily conserved program of alternative splicing: the one of neuralspecific microexons.
Microexons are tiny (3-27-nt long) exons that are critical for nervous system development and that have been implicated in autism spectrum disorders. The finding in mammals of large, tightly regulated programs of microexons came as a major surprise. It was widely assumed that very short exons were largely not accessible to the cellular machinery, as their minute size would preclude the spliceosomal interactions needed for exon definition. Therefore, these findings raised a major question: when and how in evolution did animal cells acquire the ability to recognize and splice-in microexons?
Here, we demonstrated that neural microexon programs originated in bilaterian ancestors, more than 600 million years ago, through the emergence of a distinct genomic novelty occurring in a paneukaryotic core spliceosomal gene. This genomic novelty generated an alternative isoform encoding a new protein interaction domain with a unique functional property: microexon splicing. We named this domain the “enhancer of microexons” (eMIC). We showed that the eMIC domain is necessary and sufficient for the specific inclusion of neural microexons in both vertebrates and invertebrates, and that it does so by interacting with some of the earliest factors required for spliceosomal assembly.
After its origin, the eMIC domain qualitatively expanded the regulatory toolkit of animals by enabling the recognition and subsequent selection of tiny exons that were not previously available to the splicing machineries of ancestral species. The functional importance of this mechanism is illustrated by the neurodevelopmental and behavioral defects of mouse models depleted of microexons, and also by the deregulation of this program in autistic patients.
Reference
Torres-Méndez A, Bonnal S, Marquez Y, Roth J, Iglesias M, Permanyer J, Almudí I, O’Hanlon D, Guitart T, Soller M, Gingras AC, Gebauer F, Rentzsch F, Blencowe BJB, Valcárcel J & Irimia M 2019, ‘A novel protein domain in an ancestral splicing factor drove the evolution of neural microexons’, Nature Ecol Evol, 3, 4, 691 – 701.